
最近又温习了几条法规:
1、非无菌原料药精制、烘干、粉碎、包装等生产操作的暴露环境应当按照D级洁净区的要求设置。
2、中药提取、浓缩、收膏工序敞口操作的应与其制剂配制操作区的洁净级别相适应。
(我个人一直认为按照D级区要求设置或者按照一般控制区+层流保护就足够了,说实话,真的问题不大)。
3、中药注射剂浓配前的精制工序应当至少在D级洁净区内完成
(个人认为:对于最终灭菌、非最终灭菌的中药注射剂,可以无菌过滤的和不能无菌过滤的,是否密闭操作,还是要区别对待,不能一概而论)。
4、一般情况下,普通的非无菌制剂药品的生产,生产环境也应当按照D级洁净区的要求设置。
大家一直对于“按照D级洁净区的要求设置”概念比较模糊,很多人一直在问“这样的洁净区到底怎么设置?”“D级区环境监测怎么做?”“是否要监测动态非活性粒子?”“是否要监测微生物?”总之,都没有明确的法规指南要求,需要自己风险评估。
2016年5月,WHO公布了一份指南草案,其中定义了“洁净级别从A、B、C、D延伸到E、F级”,对非无菌制剂的洁净环境微生物标准进行了规范。
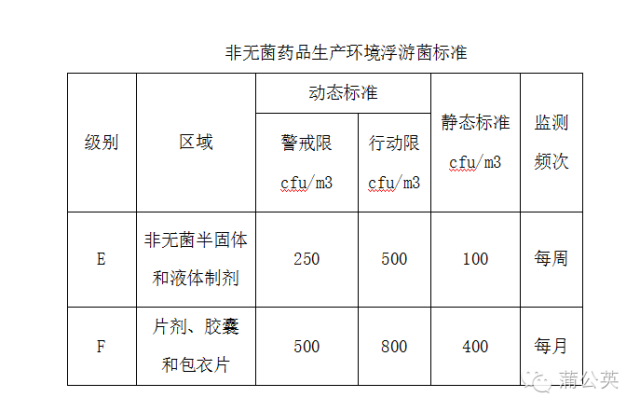
大部分非无菌制剂产品的生产环境都可以在静态条件下测试0.5um和5.0um粒子达到ISO14644-1 级别8或者D级,但是实际上对于非无菌制剂产品的生产环境,洁净度并不需要这么定义。
必须根据风险评估来决定房间所需的洁净度条件和验证的程度。
也就是说非活性粒子的可接受标准,仍然可以经过风险评估来自行确定,并不需要严格符合D级区的要求。
经过风险评估,房间洁净度和环境条件一经确定,这些条件应该被确认或者验证。
换气次数应能够满足“洁净度要求”和“自净要求”,但是D级区及EF级区“自净时间”通常没有测试的必要。
循环系统的新风比不应该被任意调整,应依据如下标准设定:
1、充足的新风以补充从设施中的泄漏和系统的排出。
2、充足的新风来满足国家建筑规范(取决于人员密度)。
3、充足的新风达到控制气味的要求。
4、充足的新风来提供必要的压差。
这时候我就在想,如果指南草案可以更清晰的规定“E级区”“F级区”的“推荐换气次数、静态非活性粒子、动态非活性粒子、静态浮游菌沉降菌表面菌、动态浮游菌沉降菌表面菌……”,那么对于绝大多数“选择困难症”患者来说,是不是更好啊?
工程师专线
135-3318-9908
联系地址:广州市南沙区南沙街进港大道577号2018房

Copyright 2020-2023 广州智造净化科技有限公司 All Rights Reserved 网站地图 网站标签 粤ICP备2022139313号



